This band structure example and associated scripts was developed by Andrew C. Pineda (AFRL),
Setting up the band structure calculation
To set up a band structure calculation, first you do a single-point SeqQuest calculation, configuring the band structure calculation using the options specified in the band structure manual. The input file to generate the CdS band structure, along a user-specified set of branches, is below.
At the conclusion of the run, the code generates two files needed to plot the band structure:
- lcao.bands – contains the band energies plotted versus distance
- lcao.bandlbls – contains the labels of the high symmetry points and their positions along this path.
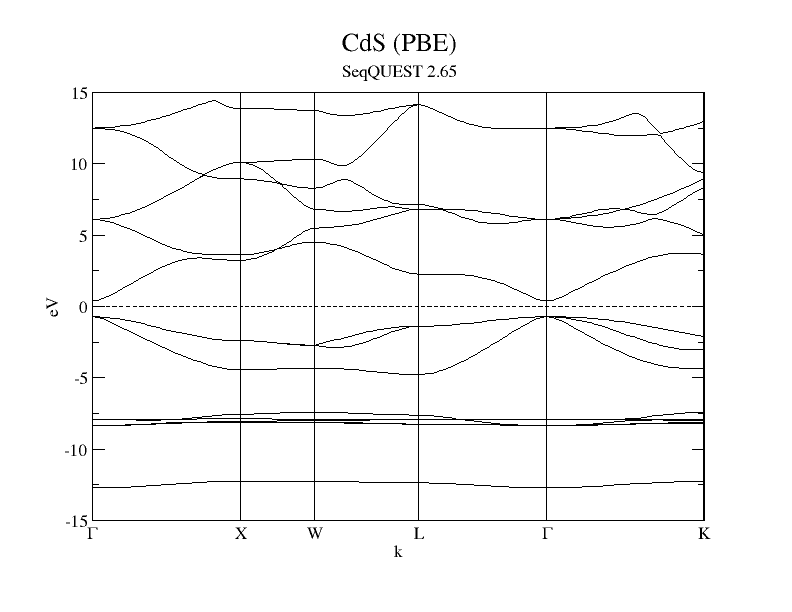
These files can be used to plot a band structure using a visualization tool such as Grace/XMGrace. A Perl script that combines these two files into a usable plot using Grace/XMGrace. This script includes options for adding labels. The figure below presents an example output from the script. The dashed line in the figure represents the Fermi level output by the code. In this figure, the Fermi level has been shifted to zero using the capabilities of Grace/XMGrace.
Example band structure for CdS
Example SeqQuest input file to generate CdS band structure
output level 4 do setup do iters no force no relax do post no cell do bands setup data notes CdS (Fd-3m)(sg 227) FINAL RELAXED CELL ENERGY = -113.1589305653 end_notes functional PBE dimension of system (0=cluster ... 3=bulk) 3 coordinates lattice primitive lattice vectors 0.0000000000 5.6252238919 5.6252238919 5.6252238919 0.0000000000 5.6252238919 5.6252238919 5.6252238919 0.0000000000 grid dimensions 36 36 36 atom types 2 atom file /home/acpineda/QUEST/atmlib-27jun07/pbe/Cd.atm atom file /home/acpineda/QUEST/atmlib-27jun07/pbe/S.atm number of atoms in unit cell 2 atom, type, position vector 1 1 0.00000000000 0.00000000000 0.00000000000 2 2 0.25000000000 0.25000000000 0.25000000000 kgrid 8 8 8 symops - for symmetry group # 227 3 definitions of symmetry operations -4 0.00000 0.00000 1.00000 0.00000 0.00000 0.00000 3 1.00000 1.00000 1.00000 0.00000 0.00000 0.00000 -2 1.00000 1.00000 0.00000 0.00000 0.00000 0.00000 end setup phase data run phase input data cutfrc - extends cutoffs for force calculations, to get better stress 5.d-2 convergence criterion 0.000010 geometry parameters gblend 0.42 gsteps 50 gconv - max force convergence criterion 0.0001 end geometry data bandstructure input bravais lattice fc2 nbranch 5 symbol labels GX XW WL LG GK end bandstructure input end of run phase data